| The CRYSTALG program locates low-energy minima by deformation (smoothing) of the potential energy surface of crystals and coupling the superbasins in the original energy surface to basins in a highly deformed energy surface. The program does not make use of any information other than the force field parameters and molecular geometry (present version of the CRYSTALG assumes the rigid geometry). As a result of the prediction all the crystal structural parameters and the number of molecules in the unit cell are obtained. |
|
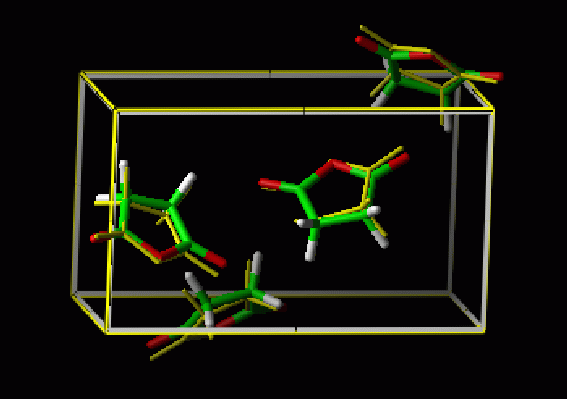
| Superposition of the experimental structure of succinic anhydride (yellow) and the lowest energy structure for the AMBER potential. |
Please complete this form if you are downloading software from the CBSU repository. We will notify you of updates and additions as they occur. You need only complete the form once.