 dziej *,
dziej *, 
 mierkiewicz *,
mierkiewicz *, 
BIOPHYSICS
Physics-based protein-structure prediction using a
hierarchical protocol based on the UNRES force field: Assessment in two blind
tests
dziej *, mierkiewicz *,
*Baker Laboratory of Chemistry and Chemical Biology, Cornell
University, Ithaca, NY 14853-1301;
Faculty of Chemistry,
University of Gda sk, Sobieskiego Str.
18, 80-952 Gdask, Poland;
Instituto de Matemßtica
Aplicada San Luis, Consejo Nacional de Investigaciones CientÝficas y TÚcnicas de
Argentina, Facultad de Ciencias FÝsico Matemßticas y Naturales, Universidad
Nacional de San Luis, EjÚrcito de los Andes 950, 5700 San Luis, Argentina;
Cornell Theory Center, Cornell
University, Ithaca, NY 14853-3801; and ÂDepartment of Chemistry,
Chungbuk National University, Cheongju, Chungbuk 361-763, Korea
sk, Sobieskiego Str.
18, 80-952 Gdask, Poland;
Instituto de Matemßtica
Aplicada San Luis, Consejo Nacional de Investigaciones CientÝficas y TÚcnicas de
Argentina, Facultad de Ciencias FÝsico Matemßticas y Naturales, Universidad
Nacional de San Luis, EjÚrcito de los Andes 950, 5700 San Luis, Argentina;
Cornell Theory Center, Cornell
University, Ithaca, NY 14853-3801; and ÂDepartment of Chemistry,
Chungbuk National University, Cheongju, Chungbuk 361-763, Korea
Contributed by H. A. Scheraga, March 31, 2005
Recent improvements in the protein-structure prediction method
developed in our laboratory, based on the thermodynamic hypothesis,
are described. The conformational space is searched extensively at
the united-residue level by using our physics-based UNRES energy
function and the conformational space annealing method of global
optimization. The lowest-energy coarse-grained structures are then
converted to an all-atom representation and energy-minimized with the
ECEPP/3 force field. The procedure was assessed in two recent blind
tests of protein-structure prediction. During the first blind test,
we predicted large fragments of  and +
and +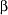 proteins [60û70 residues with C
rms deviation (rmsd) <6 ┼]. However, for
+
proteins, significant topological errors occurred despite low rmsd
values. In the second exercise, we predicted whole structures of five
proteins (two and three
+,
with sizes of 53û235 residues) with remarkably good accuracy. In
particular, for the genomic target TM0487 (a 102-residue
+
protein from Thermotoga maritima), we predicted the complete,
topologically correct structure with 7.3-┼ C
rmsd. So far this protein is the largest
+
protein predicted based solely on the amino acid sequence and a
physics-based potential-energy function and search procedure. For
target T0198, a phosphate transport system regulator PhoU from T.
maritima (a 235-residue mainly
-helical protein), we predicted
the topology of the whole six-helix bundle correctly within 8 ┼ rmsd,
except the 32 C-terminal residues, most of which form a
-hairpin. These and other
examples described in this work demonstrate significant progress in
physics-based protein-structure prediction.
proteins [60û70 residues with C
rms deviation (rmsd) <6 ┼]. However, for
+
proteins, significant topological errors occurred despite low rmsd
values. In the second exercise, we predicted whole structures of five
proteins (two and three
+,
with sizes of 53û235 residues) with remarkably good accuracy. In
particular, for the genomic target TM0487 (a 102-residue
+
protein from Thermotoga maritima), we predicted the complete,
topologically correct structure with 7.3-┼ C
rmsd. So far this protein is the largest
+
protein predicted based solely on the amino acid sequence and a
physics-based potential-energy function and search procedure. For
target T0198, a phosphate transport system regulator PhoU from T.
maritima (a 235-residue mainly
-helical protein), we predicted
the topology of the whole six-helix bundle correctly within 8 ┼ rmsd,
except the 32 C-terminal residues, most of which form a
-hairpin. These and other
examples described in this work demonstrate significant progress in
physics-based protein-structure prediction.