How do you compute similarity?
-- a trivial example
This page is based on an older tutorial by Andreas Mattern.
When we align and look at two sequences we are looking for evidence that they have diverged from a common ancestor through mutation and selection (Durbin et al pg 13). For purposes of this page, the difference between global and local is not important, but please remember such difference.
There are a number of ways in which
sequences diverge from each other:
![]() Substitution: changing one
translated product into another
Substitution: changing one
translated product into another
![]() Insertion: adding residues
to a sequence
Insertion: adding residues
to a sequence
![]() Deletion: removing residues
from a sequence
Deletion: removing residues
from a sequence
Insertions and Deletions are at times called "indels" and have the complicating factor that they may or may not be "in frame".
Let's
learn through example; Let's say
your sequence looks like this:
gattacacanttccaagacct
So,
we have a 21 nt sequence -- base composition: g=2, a=7, c=6, t=5, n=1
Let's
look at this series of variations of this sequence and the alignments it
generates:
Query: gattacacanttccaagacct
Database: gattacacanttccaagacct -->
perfect match!
Translated: DYTXPRP
Translated: DYTXPRP
--> perfect match!
7/7 = 100% similarity
Now we make one change:
Query: gattacacanttccaagacct
Database: gattacacatttccaagacct
(remember
that the n might still be a t! )
20/21 = 95% similarity
Translated: DYTXPRP
Translated: DYTFPRP (maybe
the X = F ! )
6/7 = 86% similarity
One new ( silent) mismatch!:
Query: gattacacanttccaagacct
Database: gattacacctttccaagacct
19/21 = 90% similarity
Translated: DYTXPRP
Translated: DYTFPRP --> no
change in the amino acid base!
6/7 = 86% similarity
Then we produce more silent mutations:
Query: gattacacanttccaagacct
Database: gactatacgttcccccggccc
9 mismatches!
12/21 = 57% similarity
Translated: DYTXPRP
Translated: DYTFPRP --> no
change in the aa!
6/7 = 86% similarity
See how the DNA score degrades, yet the protein score remains the same
Now we add other types of mutations...
Query: gattacacanttccaagacct
Database: gattacaca---ccaagacct --> a
3 nt gap!
Translated: DYTXPRP
Translated: DYT-PRP --> producing a 1 aa gap!
...
Query: gattacacanttccaagacct
Database: gattaca---ttccaagacct -->
an out-of-frame 3 nt gap!
Translated: DYTXPRP --> which results in a 1 aa
substitution...
Translated: DYI-PRP ...and
a 1 aa gap!
...
Query:
gattacacan-----ttccaagacct
Database: gattacacanaatgcttccaagacct --> a
5 bp insertion!
Translated: DYTXPRP --> now things are getting..
Translated: DYTXCFQD ..a little
more complicated, the reading frame has been modified!
Even in this small nucleotide sequence with very simplistic changes, you
can see how quickly things can get messy in pairwise alignment strategies.
How
could we devise a scoring scheme that would:
![]() Allow us to see the
sequences which are "pretty similar" to ours (i.e. putative
homologues)
Allow us to see the
sequences which are "pretty similar" to ours (i.e. putative
homologues)
![]() Let us ignore those sequences
which are "pretty similar" just by chance.
Let us ignore those sequences
which are "pretty similar" just by chance.
Let's look at
the simplest case: aligning two nucleotide sequences.
Using our example...
Query: gattacacanttccaagacct
--> perfect match!
Database: gattacacanttccaagacct ... 21/21 =
100% similarity
...we
could use the percent similarity score, but this can be tricky because
similarity between two sequences is not taking into account the length of the
alignment, so how do we balance between an alignment of lenght = 30 with 60%
similarity and a second alignment of lenght 200 with 60% similarity?.... ...
A short discussion , of percentage similarity (or
identity) as a single reported statistic.
...so
we see that percent similarity alone is not a good statistic to report, but as
a part of the overall report, it is very helpful to have this information. Now
look at this case ...
Query: gattacacanttccaagacct
Database: gattacaca---ccaagacct a 3 nt
gap!
… how do we deal
with insertions or deletions (indels)?
Generally,
a score is determined by:
1. giving a positive score to "matches" and a negative score to "mismatches"
2. Examining a "substitution matrix"; comprised of those scores
For arguments sake let's make a match = +2 and a mismatch = -1
The
substitution matrix would look like this:
|
|
A |
C |
T |
G |
|
A |
+2 |
-1 |
-1 |
-1 |
|
C |
-1 |
+2 |
-1 |
-1 |
|
T |
-1 |
-1 |
+2 |
-1 |
|
G |
-1 |
-1 |
-1 |
+2 |
So
without giving any values to gaps in...
Query: gattacacanttccaagacct
Database: gattacaca---ccaagacct --> a 3 nt gap!
..we
obtain:
Score
= +2+2+2+2+2+2+2+2+2-1-1-1+2+2+2+2+2+2+2+2+2 = 33
How
does that compare with:
Query: gattacacanttccaagacct
--> perfect match!
Database: gattacacanttccaagacct 21/21 = 100%
similarity
Score
= 21*2 = 42
or
with:
Query: gattacacanttccaagacct
Database: gactatacgttcccccggccc
--> 9 mismatches
Score
= +2+2-1+2+2-1+2+2-1-1+2-1+2+2-1-1+2-1+2+2-1 = 15
Pretty simple! isn't it?. But we know that this isn't really the
greatest scoring scheme in the world. The example with the gap for instance --
a larger gap (some 60 nucleotides, at least) could easily be explained by an
intron, and it should be fared better than a pair of sequences with the same
amount of mismatches; this does not apply in all gap vs. mismatch cases though,
and we might want to favor placing mismatches instead of gaps in other alignment
cases. So, depending on what we are looking for, we can also give a more or
less negative score to gaps relative to the value given to mismatches (and as
we will see later, to the extension of gaps). Modern local alignment programs (such as version 2 of BLAST
and all versions of FASTA) let’s us penalize gaps as well as modifying the
scoring matrix (some include the “-“ gap symbol as another letter whithin the
matrix).
Would the substitution scoring matrix apply for amino acids?
Also, but a substitution matrix for comparing 2 amino acid sequences
gets more complicated, as we know that some amino acids, while not identical in
chemical structure, are considered functionally conserved (i.e. D,
E, and K are all charged amino acids, while V, I, and L are all hydrophobic),
and might be functionally equivalent when replaced by members of the same
chemical properties. Remember the neutral
theory of mutation by kimura?
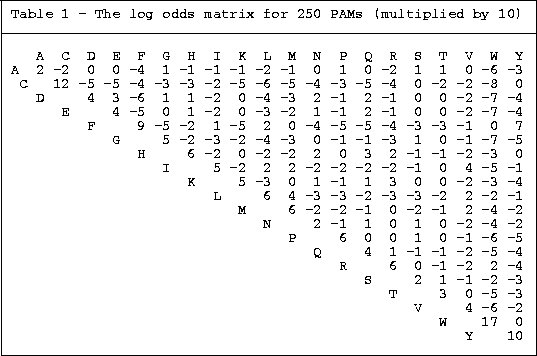
Here's an example of a commonly used
substitution matrix,
the PAM 250 matrix. Notice how some substitutions
don't have a negative score:
Now
let's return to out discussion of sequence comparisons. (close this window)