Glossary
The following list of terms have some definitions borrowed from
the glossary at NCBI, others are original, and a few have been extracted
from books, as indicated individually. The images, when not original have
been borrowed from the University of Washington at: http://www.cs.washington.edu/education/courses/590bi/98wi
.
Quick Index: A-B, C-D,
E-F,
G-H,
I-J,
K-L,
M-N,
O-P,
Q-R,
S-T,
U-Z.
Accession Number
A unique code that identifies a sequence in a database. For more advanced
users, the primary accession number is the primary key of a table in the
genbank relational database. Secundary accession numbers are other
codes that also identify the sequence but are no longer used as primary
codes. An accession number may have a version number attached
at the end if the sequence has been updated (i.e. if more sequence has
been added to it, or corrected). The accession number brings the
user to the sequence in its latest form (latest version). This is
different from gi-numbers.
Alignment
The process of lining up two or more sequences to achieve maximal levels
of identity (and conservation,
in the case of amino acid sequences) for the purpose of assessing the degree
of similarity and the possibility of homology. See this simple example
of an alignment of letters:
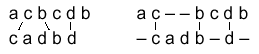
Algorithm
A fixed procedure embodied in a computer program. From Gusfield's book:
"A high level description of a mechanistic way to solve a problem or compute
a function."
ASN.1
In the NCBI context, ASN.1 is a representation of all the information that
acompanies a biological sequence (sequence of the DNA-RNA-protein molecule,
along with all other data such as authors, date, annotation, etc),
but contrary to the GENBANK format, ASN1 is suitable for parsing
by machines (manipulating the data) and less apt for human reading.
ASN1 is a standard for information transmission, and it is not limited
to biological information, in fact, this standard has long been in use
by the telecomunications industry.
Bioinformatics
The merger of biotechnology and information technology with the goal of
revealing new insights and principles in biology.
BLAST
Basic Local Alignment Search Tool. (
Altschul et al.) A sequence comparison algorithm
optimized for speed used to search sequence databases for optimal local
alignments to a query. The initial search is done for a word of length
"W" that scores at least "T" when compared to the query using a substitution
matrix. Word hits are then extended in either direction in an attempt to
generate an alignment with a score exceeding the threshold of "S". The
"T" parameter dictates the speed and sensitivity of the search. For additional
details, see one of the BLAST tutorials.
Bit score
The value S'
is derived from the raw alignment score S in which
the statistical properties of the scoring system used have been taken into
account. Because bit scores have been normalized with respect to the scoring
system, they can be used to compare alignment scores from different searches.
BLOSUM
Blocks Substitution Matrix. A substitution
matrix in which scores for each position are derived from observations
of the frequencies of substitutions in blocks of local alignments in related
proteins. Each matrix is tailored to a particular evolutionary distance.
In the BLOSUM62 matrix, for example, the alignment from which scores were
derived was created using sequences sharing no more than 62% identity.
Sequences more identical than 62% are represented by a single sequence
in the alignment so as to avoid over-weighting closely related family members.
(Henikoff
and Henikoff)
Client
A computer, or the software running on a computer, that interacts with
another computer at a remote site (server). This concept is different from
"user".
Conservation
Changes at a specific position of an amino acid or (less commonly, DNA)
sequence that preserve the physico-chemical properties of the original
residue.
Dynamic programming
Dynamic programming is a very general optimization technique that can be
applied to problems that can be subdivided into similar subproblems of
smaller size such that the solution to the larger problem can be obtained
by combining the solutions to the subproblems. These "divide and conquer"
methods are frequently used to solve alignment problems.
From http://www.mpri.lsu.edu/Chapter7.htm : "Dynamic programming
converts a large, complicated optimization problem into a series of interconnected
smaller ones, each containing only a few variables. The result is a series
of partial optimizations requiring a reduced effort to find the optimum".
Domain
A discrete portion of a protein assumed to fold independently of the rest
of the protein and possessing its own function.
DUST
A program for filtering low complexity regions from nucleic acid sequences.
E
value
Expectation value. The number of different alignents with scores equivalent
to or better than S that are expected to occur in a database search by
chance. The lower the E value, the more significant the score.
EST
Stands for "Expressed Sequence Tag", a sequence from one of the ends (either
from 5' of 3' end) of an expression clone, such as a cDNA clone from an
expression library (a snapshot of mRNAs from a tissue at a given time in
development).
By nature of current technology, an EST sequence rarely spans the full
insert sequence. The trend now is to sequence from both ends of the clone
(and when possible, to get the whole insert's sequence).
ESTs also provide marker position in a genomic map (when mapped by
recombination frequency mapping) and in a physical map (when mapped by
PCR amplyfication from or hybridization to a set of ordered large clones)
in an analogous way to STS markers; with
the advantage that ESTs are a direct link to the expressed genes.
Public EST sequences are stored in dbEST
database at Genbank.
FASTA
The first widely used algorithm for database similarity searching. The
program looks for optimal local alignments by scanning the sequence for
small matches called "words". Initially, the scores of segments in which
there are multiple word hits are calculated ("init1"). Later the scores
of several segments may be summed to generate an "initn" score. An optimized
alignment that includes gaps is shown in the output as "opt". The sensitivity
and speed of the search are inversely related and controlled by the "k-tup"
variable which specifies the size of a "word". (Pearson
and Lipman)
Filtering
Also known as Masking. The process of hiding regions
of (nucleic acid or amino acid) sequence having characteristics that frequently
lead to spurious high scores. See SEG and DUST.
Gap
A space introduced into an alignment to compensate for insertions and deletions
in one sequence relative to another. To prevent the accumulation of too
many gaps in an alignment, introduction of a gap causes the deduction of
a fixed amount (the gap score) from the alignment score. Extension of the
gap to encompass additional nucleotides or amino acid is also penalized
in the scoring of an alignment.
GI-number
A gi-number, like an accession number, is a unique
identifiers for a given sequence. A gi-number takes the user to the sequence
in its state when it was entered or modified. Every time a sequence
is updated, it maintains its accession number (with a new version number
attached) but it receives a NEW gi-number that represents its new
state. Some sequences that have been updated have a "history" of
more that one gi-number, and their different states of the sequence can
be retrieved.
Global Alignment
The alignment of two nucleic acid or protein sequences over their entire
length.
H
H is the relative entropy of the target and background residue frequencies.
(Karlin
and Altschul, 1990). H can be thought of as a measure of the average
information (in bits) available per position that distinguishes an alignment
from chance. At high values of H, short alignments can be distinguished
by chance, whereas at lower H values, a longer alignment may be necessary.
(Altschul,
1991)
Heuristic
An heuristic in a very simplified definition, is a procedure that derives
an aproximation to the real answer of a problem in a more economical or
faster way than using the more mathematically "strict" algorithm.
However, obtaining the "True" answer is not guaranteed to a 100%.
In computer science, heuristics are applied when finding the exact
solution to a problem via strict algorithms is computationally impractical.
Homology
Similarity attributed to descent from a common ancestor. Contrast this
to "homoplasy".
From another definition elsewhere
(Virginia Tech, Dept. of Biochem): One must recognize that homology does
not necessarily imply similarity. Homology has a precise definition: having
a common evolutionary origin. Thus, homology is a qualitative description
of the nature of the relationship between two or more things, and it cannot
be partial. Either there is an evolutionary relationship or there is not.
An assertion of homology usually must remain an hypothesis. Supporting
data for a homologous relationship may include sequence or three-dimensional
similarities, the relationships between which can be described in quantitative
terms. An observation of importance in homology modeling is that for a
set of proteins that are hypothesized to be homologous, their three-dimensional
structures are conserved to a greater extent than are their primary structures.
This observation has been used to generate models of proteins from homologues
with very low sequence similarities. Thus, in homology modeling, we are
attempting to develop models of an unknown from homologous proteins. These
proteins will have some measure of sequence similarity but we are relying
on the conservation of folds among homologues to guide us as well.
Homoplasy
Similarity that has evolved independently and is not indicative of common
ancestry.
HSP
High-scoring segment pair. Local alignments with no gaps that achieve one
of the top alignment scores in a given search.
Identity
The extent to which two (nucleotide or amino acid) sequences are invariant.
K
A statistical parameter used in calculating BLAST scores that can be thought
of as a natural scale for search space size. The value K is used in converting
a raw score (S) to a bit score (S').
lambda
A statistical parameter used in calculating BLAST scores that can be thought
of as a natural scale for scoring system. The value lambda is used in converting
a raw score (S) to a bit score (S').
Local Alignment
The alignment of some portion of two nucleic acid or protein sequences
Low Complexity Region (LCR)
Regions of biased composition including homopolymeric runs, short-period
repeats, and more subtle overrepresentation of one or a few residues. The
SEG
program is used to mask or filter LCRs in amino acid queries. The DUST
program is used to mask or
filter
LCRs in nucleic acid queries.
Masking
Also known as Filtering. The removal of repeated
or low complexity regions from a sequence in order to improve the sensitivity
of sequence similarity searches performed with that sequence.
Motif
A short conserved region in a protein sequence. Motifs are frequently highly
conserved parts of protein domains.
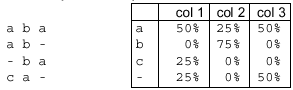
Multiple Sequence Alignment
An alignment of three or more sequences with gaps inserted in the sequences
such that residues with common structural positions and/or ancestral residues
are aligned in the same column. Clustal W is one of the most widely used
multiple sequence alignment programs. See the simple example with three
strings of letters

Optimal Alignment
An alignment of two or more sequences with the highest possible score.
Orthologous
Homologous sequences in different species that arose from a common ancestral
gene during speciation; may or may not be responsible for a similar function.
REMEMBER: separation of the two sequences due to SPECIATION event.
P
value
The probability of an alignment occurring with the score in question or
better. The p value is calculated by relating the observed alignment score,
S, to the expected distribution of HSP scores from comparisons of random
sequences of the same length and composition as the query to the database.
The most highly significant P values will be those close to 0.
P values and E values are different ways of representing
the significance of the alignment.
PAM
Percent Accepted Mutation. A unit introduced by Dayhoff et al. to quantify
the amount of evolutionary change in a protein sequence. 1.0 PAM unit,
is the amount of evolution which will change, on average, 1% of amino acids
in a protein sequence. A PAM(x) substitution matrix
is a look-up table in which scores for each amino acid substitution have
been calculated based on the frequency of that substitution in closely
related proteins that have experienced a certain amount (x) of evolutionary
divergence.
Paralogous
Homologous sequences within a single species that arose by gene duplication.
Profile
A table that lists the frequencies of each amino acid in each position
of protein sequence. Frequencies are calculated from multiple alignments
of sequences containing a domain of interest. See also PSSM.
Proteomics
Systematic analysis of protein expression of normal and diseased tissues
that involves the separation, identification and characterization of all
of the proteins in an organism.
PSI-BLAST
Position-Specific Iterative BLAST. An iterative search
using the BLAST algorithm. A profile is built after the initial search,
which is then used in subsequent searches. The process may be repeated,
if desired with new sequences found in each cycle used to refine the profile.
Details can be found in this discussion of PSI-BLAST.
(Altschul
et al.)
PSSM
Position-specific scoring matrix; see profile. The PSSM gives the log-odds
score for finding a particular matching amino acid in a target sequence.
Query
The input sequence (or other type of search term) with which all of the
entries in a database are to be compared.
Raw Score
The score of an alignment, S,
calculated as the sum of substitution and gap scores. Substitution scores
are given by a look-up table (see PAM, BLOSUM). Gap scores are typically
calculated as the sum of G, the gap opening penalty and L, the gap extension
penalty. For a gap of length n, the gap cost would be G+Ln. The choice
of gap costs, G and L is empirical, but it is customary to choose a high
value for G (10-15)and a low value for L (1-2).
Reading Frame
From Gusfield's book: "One of three places to start reading when translating
a string from the DNA alphabet into
the amino acid alphabet. If the direction of the string is also not established,
then it refers to either one of six reading frames". (Three from each of
the opposing strands).
It is necessary to maintain the frame in order to produce the right
amino acid sequence: An insertion/deletion mutation may cause the frame
to shift and have one of several consecuences, for example: 1) it may change
the original reading of the codons, generating a new amino acid sequence
or/and 2) it may introduce a new stop condon, thus creating a truncated
protein or, depending of where the mutation happens, it can completely
block translation.
SEG
A program for filtering low complexity regions in amino acid sequences.
Residues that have been masked are represented as "X" in an alignment.
SEG
filtering is performed by default in the blastp subroutine of BLAST
2.0. (Wootton
and Federhen)
Silent mutations
From Gusfield's book: "A mutation in a DNA codon that does not change the
specified amino acid. Most often, a silent mutation is in the third nucleotide
in the condon."
Similarity
The extent to which nucleotide or protein sequences are related. The extent
of similarity between two sequences can be based on percent sequence identity
and/or conservation. In BLAST similarity refers
to a positive matrix score.
Single-Pass sequence
Single pass means that a sequence has been analized on the sequencer machine only once. Generally, when the researcher is looking for the precise sequence of a clone, the sequencing reaction is served serveral times, each at a different time interval and in a separate lane of the sequencing gel. This is to produce an overlaping series of reads (all coming from the same clone and same reaction) that is used both to corroborate the sequence (by depth provided by overlap) and to extend beyond the natural length achievable in one lane by the size of the gel and by band distorsions at the bottom of the gel. It is sometimes called walking reads in a directed approach (new primers can be designed from the learned sequence to amplify and continue the walking).
Single pass is avoiding all these expensive checkups, and a reaction is loaded only one time in one lane, meaning that only one sample sequence is obtained from a given clone. Instead it relies in finding random overlaps with other clones in a shotgun sequencing approach. NOTE: In this past definition, I assume that a single lane is enough to see all four bases, as in multiplexed gel loading.
String
An ordered sequence of letters from a given alphabet. A substring
is defined as a contigous subset (portion) of a string.
Sequence Tagged Site (STS)
From Gusfield's book: "Roughly, a short DNA sequence that occurs only
once in the genome. More exactly, a pair of PCR primers within a bounded
distance, with the property that PCR succeeds using them at only one location
in the genome. STS provide markers throughout the genome, but they need
not be located in genes, in contrast to ESTs".
Substitution
The presence of a non-identical amino acid at a given position in an alignment.
If the aligned residues have similar physico-chemical properties the substitution
is said to be "conservative".
Substitution
Matrix
A substitution matrix containing values proportional to the probability
that amino acid i mutates into amino acid j for all pairs of amino acids.
such matrices are constructed by assembling a large and diverse sample
of verified pairwise alignments of amino acids. If the sample is large
enough to be statistically significant, the resulting matrices should reflect
the true probabilities of mutations occuring through a period of evolution.
Unitary Matrix
Also known as Identity Matrix. A scoring system in which only identical
characters receive a positive score.