Introductory Readings
The book by Baxevanis et al (see reference in tutorial entry page) is written for the
person who has done little or nothing in sequence analysis. Yet it is able to introduce
the reader into many important concepts and in a condensed way. There are not
that many pages to read. Durbin's book is much more advanced and requires more
attention to the details. This is the main book that we will be using for the
class after we all have the basic concepts clear and have managed to run the
programs on our own. You will not be required to read Durbin yet, but I wanted
to introduce it to you.
If you want to read a little introduction about BIOINFORMATICS per se, the NCGR site has a nice explanation.
In the following text, several words that might be new to you are described in the glossary, and in some occassions a direct link to the glossary has been set. however, the glossary by itself is rich in definitions that may not be used in these webpages, but may appear on the book readings or are useful to learn. We encourage you to consult the glossary for those extra definitions, and we invite you to follow the hyperlinks in the glossary. These are links to external pages that complement the short description from the glossary and may provide good extra explanations or graphical explanations.
Internet resources (optional)
Please begin by reading Chapter 1 of Baxevanis et al
("The internet and the Biologist") if you feel that you need to
strenghten your computer and internet skills. Pay attention to the headings
related to "Electronic Mail" (p.4), "File Transfer
Protocol" (p.7) and the "World Wide Web" (p.8). For
most readers, ch.1 will not be necessary, however.
In addition, there are good resources in the web that teach
you how to navigate and how to do power searches for just about anything you
want in the internet. A good example is this page from the library of UC at
Berkeley: http://www.lib.berkeley.edu/TeachingLib/Guides/Internet/FindInfo.html
Sequence databases
There is a complete listing of specialized databases on the 2000 biological databases issue of Nucleic Acids Research. This issue is available online, but I suggest you read this text before visiting that page.
Definitions
Databases, like the name implies, are a collection of data. In this case, we are talking about a collection of information related to biological sequences (biopolymers). Those sequences may be from DNA molecules, RNA molecules, proteins and all variants of them (like mRNA, tRNA, htRNA, DNA primers and oligos, special peptides, etc). In general, databases are specialized for the two major types of macromolecules, separating the nucleic acids in one category, and proteins in the other. There are also other categories in which certain database specialize further, for example, the UniVec database (from the Genbank set), which specializes in the sequences from vector molecules adapters and linkers used by molecular biologists for cloning nucleic acids. Another example of a specialized database may be GCRdb, that holds sequence data of the “G protein-coupled receptor” class of proteins.
The respective creators maintain several of the publicly available databases, particularly the very specialized ones (special purpose oriented), but there are also large generic databases as well as centralized centers for storage, maintenance, collection and distribution of these generic databases. Possibly the most well-known are the national centers such as the EMBL, which is the repository center for the European Community, the DDBJ which is the national center for Japan, and for the U.S the NCBI, the principal center and the recently created NCGR, a competitor which also houses public data. There are also private agencies that store public databases such as TIGR. In addition, several research centers and some industries provide access to databases funded by them, like the Merck/NCI Human EST database, maintained in the Washignton University School of Medicine in St.Lois, MO. So there are plenty of public databases out there to explore.
How do we obtain the sequences that fill up the databases (or the ones that we will use as query)? If you wish to read a fast reminder of how scientists collect sequences of nucleic acids, check this page, but this is entirely optional. Protein sequencing uses a different methodology (in fact, there are serveral methodologies). You can also check these in a modern biochemistry book.
Once you obtain a sequence from your experiments, perhaps the very first places to look for others resembling yours are the generic databases. It is useful to compare your sequence with a list of known ones, because this is the fastest way to infere some origin or function to yours (you will probably need to do some comfirmation experiments later).
As it was said before, the most recognized centers for databases are the NCBI (maintains Genbank), EMBL and DDJB. All three centers share their data and they update each other everynight, so for most cases you only need to visit the one which is closest to you geographically. For our case, Genbank fills most of our needs; there are a few instances when we may want to go outside, to visit the specific databases, or to explore database searching programs not offered by the NCBI.
Let’s review some of the most used databases.
Genbank. Nucleic acids dababases
![]() nr (or “nt” when you download it)
nr (or “nt” when you download it)
Non-Redundant database, from GenBank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS, or HTGS sequences). This is the most generic of the nucleic acid databases. For the purpose of archiving and downloading, it is subdivided in taxonomic “divisions” but the categories of subdivisions are not always at the same level: sometimes at “kingdom” level such as the green plants division, sometimes at “class” level such as the mammalia division, or not defined such as the invertebrate division (just like saying “Metazoa” But Not “vertebrata”).
The user can use taxonomic terms when querying this non redundant database to restrict the searches (or the retrieval of sequences) to the desired group of organisms. The database is intimately linked to a taxonomy database for these purposes; as we will see later, all genbank records have a field for the taxonomy of the source of the sequences.
![]() month
month
All new or revised GenBank+EMBL+DDBJ+PDB sequences released in the last 30 days.
![]() dbest
dbest
Non-redundant Database of GenBank+EMBL+DDBJ EST Divisions. This is a database of Expressed Sequence Tags; short, single pass reads of cDNA (mRNA) sequences. Also includes cDNA sequences from differential display experiments and RACE experiments. By nature of their origin (single-pass), they are considered of poor quality yet they are VERY useful in a variety of topics, such as finding codign regions in unknown genomic sequences, proving alternative forms of splicing of genes, or as alternatives to STS physical/genetic mapping. Do not trust the definition lines of many ESTs for which homology assumptions have been described there since these assupmtions were obtained from automated assesments of database searches and are rarely updated. (Williams. G. in Bishop et al.).
ESTs represent more than half of the size of NCBI’s databases. Human and mouse are the most represented organisms today. The databases however contain a fair to large amount of non-quantified redundancy, which is useful for finding overlaps and estimating longer consensi sequences (by clustering techniques), which in turn upgrades the quality level of the information contained in the raw sequences. ESTs are used to generate unigene estimates (See gene indices later in this section).
![]() dbsts
dbsts
Non-redundant Database of GenBank+EMBL+DDBJ STS. This is a database of Sequence Tagged Sites; short sequences that are unique in the genome, used to generate mapping landmarks. It contains detailed information about sequencing primers and PCR conditions as well as physical map location. Helpful in both physical and genetic mapping. A significant sequence similarity match against an STS marker will help to locate your sequence against the physical map. Please remember that STS are originated from genomic sequence, and may be located inside or far from genes.
![]() gss
gss
Genomic Survey sequences such as single-pass genomic data (like large clone ends. i.e. cosmid/BAC/YAC ends) , exon-trapped sequences, and Alu PCR sequences. Since they are single pass, they are not appropiate for inclusion in organism specific databases (requiring a minimum error rate). These sequences are useful in the creation of physical maps (By means of overlapping clones) for posterior full sequencing.
Others:
![]() htgs
htgs
Unfinished High Throughput Genomic Sequences (htgs) in phases 0, 1 and 2. (finished, phase 3 HTG sequences are in nr) . These sequences are released by public high throughput genome efforts within 24 hours of the sequencing, and get updated with automated annotation progresivelly, until full completion of sequence assembly when they get moved to the nr database under the respective taxonomic division. They are composed of very long genomic sequences and have an assigned position in the genome they belong to. Because of the continous updating, these sequences are not very stable. Entries in this database are mostly from Human, nematode and Arabidopsis, since they are unfinished genomes.
![]() Full genome
databases
Full genome
databases
One for each of E.coli, yeast, and Drosophila complete genomes as well as a plethora of prokariote genomes, released by various sequencing consortiums. The nematode genome (C.elegans) is not included because their authors state that it is still in progress. Other eukariote organisms are in working phases as well and they are included in the htgs division.
![]() mito
mito
Database of mitochondrial sequences from several organisms. Do not confuse them with nuclear sequences that express proteins that are targeted to the mitochondria.
![]() epd
epd
From Eukaryotic Promotor Database found on the web at http://www.genome.ad.jp/dbget-bin/www_bfind?epd
![]() dbSNP
dbSNP
This is a database of Single Nucleotide Polymorphisms, small-scale
insertions/deletions, polymorphic repetitive elements, and microsatellite
variation.
![]() patent
patent
Contains sequences mentioned in patents in the diverse patent offices (National and International). Sequences from patents as far as 1960. (Williams. G. in: Bishop et al.).
Genbank. Protein databases
It may be a good moment to remind the student that since nucleic acid sequencing is much cheaper than protein sequencing (at least, currently), many of the protein sequences in most of the databases are in fact predicted amino acid chains derived from algorithmic (conceptual) translations from the DNA/RNA source.
The only NCBI owned database of proteins is that of translations made from the Genbank nucleotide sequence. Other sequences derived from external sources are appended into NCBI’s protein data. Perhaps the most famous protein database external to NCBI’s databases is SWISS-PROT, but it also has a great portion of translated sequences. Because of this, sometimes a frame error in the original nucleic acid sequence may be translated into a fake (or partially wrong) protein sequence. KEEP THIS IN MIND, and be careful with protein sequences. About 5 to 10% of sequences in SWISS-PROT (which is considered of the highest quality of databases), have been found to contain frame-shift errors (Williams G. in Bishop et al.).
![]() nr
nr
All non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF
![]() month
month
All new or revised GenBank CDS translation+PDB+SwissProt+PIR+PRF released in the last 30 days.
![]() swiss-prot
swiss-prot
The last major release of the SWISS-PROT protein sequence database (no updates) is included into Genbank's protein data. The original SwissProt is housed in the EBI (EMBL) an is produced in collaboration with the University of Geneva. Data in SWISS-PROT is manually curated and is derived from several sources: translations from DNA sequences from the EMBL nucleotide sequence database, extracted from the literature or submitted directly by researchers. It contains high quality annotations and is non-redundant. Its sequences contain cross references to other databases such as the nucleotide db at EMBL, the PROSITE pattern database, the PDB protein structure database and others.
![]() pdb
pdb
Sequences derived from the 3-dimensional structure Brookhaven Protein Data Bank. These are more reliable in the sense that their biological function has been confirmed biochemically and their 3D structure is known.
![]() Full genome
databases:
Full genome
databases:
One for each of E.coli, yeast, and Drosophila (from Celera + U.Berkeley sequence). All derived and known proteins from their genomes.
Other Protein Databases (but not included in Genbank)
Blocks , Prints à (identify local matches to your query),
ProDom, Pfam, ProfileScan à (identify global matches to your query).
![]() TrEMBL
TrEMBL
This database is a complement to SWISS-PROT. All the translations from coding sequences in EMBL that are not yet integrated in the high quality SWISS-PROT are in TrEMBL and contain computer generated annotations.
It may be considered as a preliminary SWISS-PROT. It is split into two sections, SP-TREMBL, containing sequences which would eventually get incorporated into SWISS-PROT, and RE-TREMBL that contains sequences not to be included in SWISS-PROT.
SP-TREMBL sequences have been assigned a SWISS-PROT accession numbers, while RE-TREMBL sequences do not have an accesion number at all. This last database contains sequences from immunoglobulins and T-cell receptors, synthethic sequences, patent application sequences, small fragments and translated coding sequences where there is a belief that the peptides are not real (from ineffective ORFs). (Williams G. in Bishop et al.).
![]() Prosite
Prosite
Dictionary of protein sites and patterns at Expasy. Stores protein active sites, patterns and profiles that help to identify to which known family of proteins your sequence belogs to. (Williams G. in Bishop et al.).
![]() Blocks
Blocks
Blocks is a database of protein motifs that is derived from the Prosite database, and attempts to represent the most conserved of the motifs from groups of related proteins regardless of whether there is an assigned biological funtion to them. In contrast, the profiles from motifs listed in Prosite have a funtional basis. A Block is then a short contigous interval in a multiple alignment of amino acid sequences.
![]() Prints
Prints
Protein Motif Fingerprint Database. “A database of groups of conserved motifs used to characterize a protein family, they can encode protein folds and functionality more flexibly and powerfully than can single motifs.” (Williams G. in Bishop et al.).
![]() Pfam
Pfam
Protein Domain Families. "A high-quality comprehensive collection of protein domain families."; (Williams G. in Bishop et al.).
![]() ProDom
ProDom
"A protein domain database produced automatically from the SWISS-PROT database". (Williams G. in Bishop et al.).
Gene indices
![]() Unigene set
from NCBI (Human gene index and others)
Unigene set
from NCBI (Human gene index and others)
The “Unigene set”, an approach developed by scientists at NCBI, represents the first attempt to cluster human EST information in order to reduce complexity and redundancy of the EST database (Schuler, Boguski et al. 1996). It originally took the 3’ end human ESTs (which were the vast majority) and clustered them with mRNA sequences extracted from Genbank.
5’end sequences were added to the clusters using clone name information in the database (i.e., information derived from the annotation of ESTs, where some of them indicate that had been derived from the opposite ends of the same cDNA clones).
Prior to clustering, all ESTs were checked to remove contaminant sequence from vector and primers. They also were filtered with the " Dust" program from NCBI to mask low complexity regions. Clustering was based on similarity and was done with a modified Smith-Waterman algorithm or with BLAST. After reporting the members for every cluster, no attempt was made to derive consensus sequences.
Unigene sets have been assembled for humans, mouse, rat and the Zebra fish and are available at http://www.ncbi.nlm.nih.gov/UniGene/
![]() TIGR index
for humans and several other organisms
TIGR index
for humans and several other organisms
The Institute for Genome Research (TIGR) has compiled its own version of a gene index for humans and several other species. For the first incarnation of their gene indices, the institute used a set of in-house software for clustering and assembly as well as the public domain programs BLAST and FASTA.
First, a set of annotated full-length gene sequences was collected and their CDS feature annotations (annotations about the coding sequence) were parsed to construct a database of expressed gene sequences where redundant sequences are eliminated. The annotations were checked for consistency. This first step constitutes the EGAD database.
Then they downloaded the ESTs for each of the species under study from NCBI and cleaned them from untrimmed vector, low quality and polyA/polyT sequences.
With the aid of the THC_BUILD program, TIGR created index groups based on an all-way pairwise sequence similarity of the joined EGAD and clean EST databases (with aid of FASTA and BLAST programs) and then stored the overlap results in a relational database. It used the program CLUB to query the newly formed relational database and by means of transitivity relationships of similarity, found all similar sequences producing clusters of overlapping sequences. The criteria used to cluster was that two sequences must overlap on at least 40 nucleotides with a minimum of 95% identity and be fewer than 20 nucleotides of unmatched sequence at the ends. Then every cluster was assembled separately using the program TIGR_Assembler (Sutton,White et al. 1995) with high stringency settings to produce high quality consensi. Assembly may produce more than one consensus per cluster and rejects all detected chimeras.
Those clusters that contained a representative from the EGAD database (the database that contains known and annotated mRNAs) were assigned the function described in that database. This method is considered to be very strict in the clustering, and does not keep track of possible mRNA variants that get lost in separate tentative contigs. It also results in a more fragmented representation of the EST dataset than other clustering methods.
The second and most recent incarnation (Quackenbush, Liang et al. 2000) of the gene index for some crop plants such as rice and Arabidopsis (OGI 3.0 released in December 1999) has a few modifications. For instance, it no longer used TIGR_Assembler as the assembly program but instead used CAP3 (Huang and Madan 1999) but TIGR's gene index still does not reference possible splice variants.
![]() SAMBI
Sanigene
SAMBI
Sanigene
The Human Sanigene database (Miller, Hide et al. 1999; Miller, Christoffels et al. 1999) is yet a third view of the current information about the human “transcriptome”. It is somehow an intermediate between the Unigene and TIGR human gene index in terms of the philosophy for clustering. Like NCBI’s Unigene approach in the beginning, SAMBI provides a gene index only for humans and there are no indications of future plans to construct one for other species (like plants) as TIGR did. The set of programs used to cluster include the assembler PHRAP developed at Washington State University (Green 1999) and the in-house software collection called “STACK PACK”.
The STACK PACK system breaks down the EST database into bins according to the origin of the mRNA used to make the libraries (tissue and organ, disease status), then does the cleanup of the sequences (removal of vector, masking of low-complexity regions, etc) and performs a loose first stage clustering (which we call preclustering) by sequence similarity with the d2-cluster algorithm (Burke, Davison et al. 1999). This similarity algorithm uses a word pattern matching (similar to a lookup in a dictionary) rather than local sequence alignment, and this makes it faster than BLAST for finding similar strings, however, it does not attempt to construct alignments. This loose clustering is claimed to increase the possibility of retaining splice variants.
The loose clusters are assembled with PHRAP to produce consensi sequences for each cluster and then each consensus is checked for consistency of the members with the derived consensus using the CRAW and Contigproc programs from the Stack Pack. A final join of consensi sequences is done via clone linking (by means of annotation of the EST dataset) in the cases where ESTs belonging to a common sequenced clone failed to join by similarity (probably due to lack of overlap). This stage of clone linking is left to the end because it relies on annotation from the ESTs, which can be erroneous. By leaving the inclusion of possible error to the end, they avoid propagating them.
The Stack Pack system is available in Europe at the EMBL, in SouthAfrica at SAMBI (South African National Bioinformatics Institute, the original source) and in the United States at the NCGR.
![]() More
expression databases
More
expression databases
The 2000 biological databases issue of NAR has a listing of specialized expression databases. (Part of the big listing mentioned at the beginning of this chapter).
Metabolic Pathways
One of the best metabolic pathways databases is the Kyoto Encyclopedia of Genes and Genomes (KEGG) in Japan.
They also have a listing with pointers to other metabolic databases.
Synopsis
Nature Genetics has short review about the utility of certain databases.
Just as a note, keep in mind that databases of biological sequences are VERY PRONE to contain errors. In particular, large databases where many researchers have contributed sequences and released them "publicly" may contain contaminations from vectors (of host cell genomic DNA).
In the case of annotation, be warned that this is still a rudimentary process, and that we are learning it as we go along. It may happen that function assignation is derived from a single sequence (A to B) and that a chain of propagated annotations if formed from here, for instance, a sequence D that has been annotated as being of X function based on its similarity to C, which in turn was annotated based on B which was based on A, only to find later that the original sequence A has been updated later because it contained an error. This of course was never corrected in sequences B,C and D.
How to get sequences: Entrez (from point of view of Web search engines)
A generalization of "find" functions in web browsers and search engines
Have you ever used the "find" function from your word processor of browser? If you haven't, open the "Edit|Find" menu on the top of your browser, and in the little window where it asks you what to search for, type:
"Let's find THIS exact phrase"
Type exactly what is there between the quotes, the find little program should bring you back to the previous paragraph. Did it work?, well, the find little program is not much different from any other "find" algorithm used to find a piece of text in any computer file, be it a word processor text or an item in a database or a webpage indexed in a web searching engine such as Yahoo.
Searching algorithms do something that is called "String matching". What you use to search (the query phrase above) is the "pattern" of letters to match against a text file or list of items in an index.
An index is a list of words used in a text, generally ordered alphabetically. The purpose of the index, as you may already know, is to speed up searches of specific keywords in a book (a much faster alternative than browsing the whole text of the book looking for your keyword); once you find your keyword in the index, you have a pointer to the page of the book where the keyword appears. Databases do the same trick. Generally, an index is constructed a priory for every field in the database tables that are probably going to be searched by users. This way the searches are much faster.
You can make more complicated searches, for instance in the above example, you had to type the exact phrase in order to pinpoint on the exact place in the text, but what about when you don't have ALL the information about what you want to search for? In such cases you can use more generic information and retrieve a list of possible matches hoping that your target will be contained within the answer set. In the example above, the find program matched exactly five consecutive words and in the order that you specified. If you repeated your search but changing the order of the words you probably would not find the paragraph as you did before. Other type of searches (but not supported by your browser) allow you to find your words regardless of the order in which they appear, evenmore, some would allow you to look for the words even if they are not consecutive, but within certain distance of of each other that you determine (a sort of "limit").
Let's think of a more biological oriented example. Let's say that your lab is interested in two distinct families of proteins: proteases and cytochromes.
In the following diagram, see a representation of the two sets of proteins in the outer circles and the representation of the protein sequences of mouse and human in the rectangles in between them.
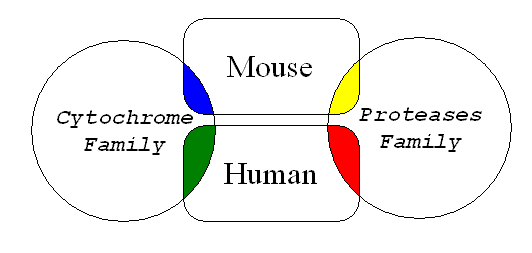
The blue color depicts the set of mouse Cytochrome related proteins (Subunits and related proteins) available in the databases, while the green color represents the human version of the Cytochrome related proteins. Likewise, Yellow color represents the collection of mouse derived proteases which are available in the databases and the red color represents the human proteases in the same databases.
A good searching engine in a database (and most modern ones are) will let you make advanced queries. For instace, you could ask the database search engine to list you all human proteases and human Cytochrome related proteins. Your query in schematic English would be something like this: find -> human and (Cytochrome or protease). The search engine should retrieve you all green and red sequences. Just remember your high school classes in Set Theory; "AND" means the intersection between two sets, and "OR" means the union.
Or, you could make a different question, such as, find all mouse cytochrome and human cytochrome related proteins: find -> cytochrome and (human or mouse). The answer should contain all blue and green sequences. This is because the search engine matches your keyword strings (cytochrome, human, mouse) with the specified "boolean" relationships ( and, or ) in the tables of the database (just like the little "find" exercise from above).
What about this one: find all sequences from human and mouse corresponding to cytochrome and protease related proteins: find -> (mouse or human) and (cytochrome or protease). The answer should give you all blue, green, yellow and red sequences.
A tricky question is this: Could you formulate a query like this one: find -> (human and mouse) ?. This query should be interpreted as: "Find one or more sequences that belong to humans and mouse", and according to the diagram above the result should be empty.
Basic searching
& refined searching in Entrez (boolean expressions, limits, etc.)
Entrez is an interface (a view) of the databases stored at the NCBI, including the already mentioned Genbank. This interface allows you to search their databases in a similar way to the little "find" program that you used a couple of paragraphs ago. In Entrez you can retrieve the objects stored in Genbank (biological sequences) just like you would retrieve any other object in other databases.
The process is analogous to going to your local library and searching for a book you wish to read. You first search for your book in the card files of a catalog using information regarding such book, like the name of the author, the publisher, the year of printing and the title of the book. Let's say you prefer to use the author's name. You start by matching the first letter of the author name with those in the ordered list of authors in the cards, then you match the second letter, the third, etc. Finally you find an exact match or something that closely resembles it. If you find an exact match, the card will provide you with a location of the book in the shelfs, (like the "page pointer" in the index of a book). If you don't find an exact match but a close resemblance, you will need to use more information about the book to search, like the tittle or subject (perhaps you are misspelling the author's name). You might even settle for another book of the same subject if your wished-for book is unavailable in that library.
Please read the Introduction to the Entrez query system, and When you are done with the reading of that webpage, please also read chapter 5 “Information Retrieval from Biological Databases”, starting at page p.98 of Baxevanis et al. book. Read the introduction but you do NOT need to read pages p.99-100, which deal with the e-mail version of Entrez. Instead skip to page p.101 containing “Integrated Information Retrieval: The Entrez system” heading and keep reading until the end of the chapter.
Homology vs. similarity
Strictly, homology refers to the situation where nucleic acid or protein sequences have a common evolutionary origin. Often used loosely to indicate that sequences are very similar. Sequence similarity is observable; homology is an hypothesis based on the observation of similarity.
We are interested in finding sequences which are homologous
to our search sequence, but we'll have to settle for sequences which share
similarity to our search sequence.
In general, two sequences are homologous if their sequences are likely to be offsprings of a common ancestor sequence. BUT we rarely have ancestral DNA to play with, SO homology is INFERRED from similarity of existing sequences – more similar more likely the homology assumption.
You can not say "percent homology" -- saying "two sequences are 25% homologous" is like saying that a “friend of yours is 25% pregnant". It just doesn't make sense! (from Andreas webpage). You will see this little mistake all over, even on some tutorials at the NCBI. If you catch this little error, remember that the author probably meant “percent similarity”. :)
In Evolution theory, the concept of homology (applied to two sequences; i.e. two pieces of DNA containing a similar gene) can be subdivided in two types, depending on the evolutionary origin of the homology relationship:
These two concepts have nice definitions, but in practice, is very hard to assign two similar sequences two one or the other type of homology hypotheses. For a graphical explanation of the two concepts, check this graph.
One of the goals of sequence comparison is to determine whether the sequences have sufficient similarity to justify the inference of homology (Schuler 1998) (it is possible to have another goal in mind, such as plain structural similarity, without concerns about homology) and for cases when homology is already suspected, sequence comparisons may be undertaken to derive a phylogenetic tree. The most common way of comparing two or more sequences of proteins or DNA molecules is by aligning them.This method can tell the researcher which pairs of residues from two sequences (either nucleotides or amino acids) are identical or at least similar, in the context of their relative position in the sequences.
Pairwise and Multiple Alignment
If only two sequences are compared, the method is a pairwise alignment and if more than two are compared, then it is a multiple alignment. However, a multiple alignment program may use as part of its procedure the iteration of pairwise alignments for every possible pair of sequences in the experiment.
The number of possible ways to line up the letters in a pair of sequences depends on the sum of their lengths (thus in multiple alignments, the number of sequences also increments the number of possible alignments). There is in most cases one alignment that can be considered the best (optimal), but the algorithm used to solve this problem needs to find it among a large number of non-optimal alignments.
The first optimal alignment methods for biological sequences were called global, because the algorithms that compared the sequences attempted to find the best alignment over their entire length. This global method is used when there is reason to believe that the sequences compared share a common origin over their complete lengths (Ostell and Kans 1998) and this belief comes from experimental evidence of their function. Most multiple aligment programs, such as "ClustalW" are based on global alignment.
The algorithm from Needleman&Wunsch (Needleman and Wunsch 1970) is one solution to the global alignment question. It uses a succesful problem solving technique called dynamic programming. Since then, others have inproved on it; for instance, Gotoh described an enhanced version of this algorithm (Gotoh 1982). We will see in class what this algorithm does, but in essence, it works by assigning a score (a value that measures qualilty) to each of the possible alignments between two sequences and reporting the one with the best score. The score for each possible alignment is calculated by summing the incremental contributions of each step while the alignment is being constructed. Positive scores are assigned for every match of identical residues in the alignment and negative scores for every mismatch (residue substitution) or gap.The optimal global alignment must extend from beginning to the end in a pair of aligned sequences (Schuler 1998).
The programs for searching or "querying" unknown sequences against a database of stored sequences or for searching for possible similarities between two given sequences without prior knowledge of homology are based on local alignment algorithms rather than global alignment algorithms.They are called local, because they concentrate on comparing subregions within the sequences as opposed to trying to align them over their full length, but they are based on the same generic concept as the global alignment.
The classical algorithm example for local alignment is the one by Smith&Waterman (Smith and Waterman 1981). In this algorithm (also based on dynamic programming), an alignment between two sequences may start and may end at any point internally, and need not reach the edges. There can be many individual regions of good alignment surrounded by regions of very poor alignment.
Modifications made to the algorithm allows it to report those alignments that are good above an statistical threshold set by the user (you), meaning that it can report more than one significant optimal alignment if there are several (between a couple of sequences), instead of reporting just the best one. Other alignment algorithms (such as BLAST) refer to one of these optimally aligned portions of a pair of sequences as a High Scoring Pair or HSP. An MSP or Maximal segment Pair is that HSP with the highest score.
The local alignment is useful when comparing sequences that are from different sizes, or when the user expects to find results that are “contained” within the query sequence; i.e., a large clone, such as a BAC sequence against a database of known genes; or when two sequences compared no longer align in the global fashion, but still conserve domains of similarity; i.e., the case of two proteins sharing a similar domain. It is also useful for discovering similarities between sequences of different biological origin; for example, a search for which the query is a processed mRNA sequence and the target database is a list of genomic sequences (possibly containing introns).
In database searches, the query sequence is compared with each of the members in the database using local alignment. The problem with full “dynamic programming” algorithms such as the Smith&Waterman algorithm (or S&W) is that they are slow (or computationally expensive) for very large sequences or for large databases because they explore the entire space of possible alignments in each pairwise comparison. A database such as dbEST release 010700 (January 2000) containing 3,458,198 sequences had around 1,320,000,000 nucleotide bases (a rounded estimate). The comparison of a single sequence of length 300bp against this database requires creating a matrix with 3.96x1011 cells (equal to the product of the sequence length and the database length).This can easily take several hours to compute in a standard modern workstation, making the optimal dynamic algorithm methods impractical.
Because of this, programs that use heuristics have been written, and shown to be much faster than Smith-Waterman (getting the result in seconds or minutes). They were designed to reduce the number of alignments pursued per pairwise comparison while still obtaining the high scoring ones. These programs, by “cutting some corners” can be tens of times faster but run the risk of missing some important true alignments. Among the many existing heuristic programs of database searching two became widely accepted, FASTA and BLAST, although BLAST became the most popular. The decition of which heristic performs better is left to each individual user and the kind of searches needed (sometimes one package is better for some tasks, while the other is best for others, and we will learn and practice with both of them in class).
The BLAST package has several programs to compare nucleotides or proteins against a pre-made nucleotide or protein database.The program may run in a distant centralized location that offers the service of database search such as the NCBI (the developers and distributors of the BLAST programs). The programs can also run locally and be used to search locally developed databases or the same databases that NCBI distributes, such as Genbank and dbEST.
The original FASTA service is at the Univesity of Virginia. This package also contains functions that allow to search with DNA or Protein sequence against a nucleotide or protein database and it can also be run directly on the servers, or it can be downloaded and run locally.
Book Reading:
Pages 145-150.
Please read about pairwise (both global and local) alignment in the following sections from chapter 7 (page 145) from Baxevanis et al Book: Start at page 145, reading the "Introduction", continue with "the evolutionary bases of sequence alignment", and "the modular nature of proteins" until its end at page 150.
Reminder of Statistics
You are expected to know an intermediate level of statistics. You will understand this class better if you already took the classes required for all graduate students: Stats 601 and 602, from the biometry department. In particular, you should understand the concepts of independence, expectation values, basic probability distributions, joint and conditional probability and Bayes's theorem, etc. However, if you are a little rusty on your math, we recommend that you check the following page (A VERY basic tutorial) http://www.robertniles.com/stats/ and then go ahead to the library to check your statistics book to remember all the rest.
Alternativelly, there is material on the internet that will help to remember the statistical concepts that you need for this class. We recommend that you also visit these pages:
![]() The Statistics Refresher Map at George Mason University: Follow the probablility (red) line through each of the four topics until you complete all of them including the self-tests. (Basic probability, random variables, expectations, and distributions).
The Statistics Refresher Map at George Mason University: Follow the probablility (red) line through each of the four topics until you complete all of them including the self-tests. (Basic probability, random variables, expectations, and distributions).
![]() You also need to understand Bayes's Theorem. Since this is not included in the previous webpage, we have provided you with another link to a site in Los Alamos National Labs that explains the theorem with a very good example. Go to introduction to bayesian inference. You only need to read this page, so please avoid any links within that site. Extra, optional reading: After that excellent introduction, if you want to explore more on how Bayesians approach probability, there is a good paper by Pr.Tom Loredo, now at Cornell's astronomy department, with an introducion to bayesian analysis, a little on history and how it can be applied. A second paper that introduces Bayesian principles (Jefferys W.H and Berger J.O. 1992. Okham's razor and Bayesian Analysis. American Scientist 80:64-72) is available on reserve in Mann Library.
You also need to understand Bayes's Theorem. Since this is not included in the previous webpage, we have provided you with another link to a site in Los Alamos National Labs that explains the theorem with a very good example. Go to introduction to bayesian inference. You only need to read this page, so please avoid any links within that site. Extra, optional reading: After that excellent introduction, if you want to explore more on how Bayesians approach probability, there is a good paper by Pr.Tom Loredo, now at Cornell's astronomy department, with an introducion to bayesian analysis, a little on history and how it can be applied. A second paper that introduces Bayesian principles (Jefferys W.H and Berger J.O. 1992. Okham's razor and Bayesian Analysis. American Scientist 80:64-72) is available on reserve in Mann Library.
There is also a reminder in statistics in chapter 11 of Durbin's et al book. This is the book that will be used later for the class. However, this chapter 11 is rather a little complex for those who are rusty, please do the recommended homework from above and read the "Introduction" of this book before you check this extra chapter 11. If you can understand what is there, then your level of statistics is more than sufficient for this class.
Warning: Please make sure your level of statistics is more than just the basics. Otherwise you will have a hard time understanding the lectures.
Repetitive and low complexity regions
What are they?
A common feature of medium to large genomes is the presence of repetitive regions in the chromosomes. Such genome's DNA can be partitioned in at least three categories: single copy DNA, moderatedly repetitive DNA and highly repetitive DNA, as defined by a DNA denaturing/re-association kinetics (or "Cot") curve. More often than not in the past, we focused in the single to low copy regions of the DNA because this is where most of the genes lie, but there are also some genes that are repetitive (or located in repetitive regions) and therefore we must look at those sections of the genome as well. (see these sets of slides: 1, 2, 3, if you don't remember this).
These repetitive regions may vary in several aspects, such as the base composition (very elemental or very biased in terms of which bases occur in such sequences), the length of the smallest common word (the repeated motif) and the times that such motif is repeated both in tandem and throughout the genome. Minisatellites, Microsatellites, Transposable elements, are all features that make up the repetitive regions of the genome. In some instances, these repetitive elements (not necessarily in tandem) can be around or inside genes.
Low complexity regions in the DNA are biased in the base composition, for example, the region may be exceedingly rich on A and T bases (or any other combination), or even a repetition of a single base, such as the poly-A tails of eukariote mRNAs.
Why do they matter?
Repetitive sequence and low complexity sequence in a query sequence (for instance, when searching Genbank) will interfere with the searching in the database and will probably retrieve unwanted matches.
For instance, if your query sequence is a messenger RNA, and it contains a polyA tail (you forgot to remove it), this polyA tail, if left unfiltered will retrieve many other messager RNAs in the database that also contain polyA tails. The chance that those random matches to other polyA tails is of any biological significance (other than just being that, a polyA tail added to ALL mRNAS) is basically null. If the scores caused by matching those repetive or low complexity sequences is sufficiently high, they can override any true matches found in the database.
Book Reading:
Pages 166-169.
Please read the headings "low-complexity regions", and "repetitive elements" of chapter 7 of Baxevanis et al book, starting at page 166 and finishing at page 169.
Masking to improve biological fidelity of results
To filter or mask the low complexity regions in a DNA sequence use the ' Dust' program. This is also available as an option that you can turn on when performing DNA searches with the BLAST program (in any of its flavors local or web based). The protein counterpart is ' SEG'. There are other programs that have similar functionality when the tasks needed do not involve BLAST. RepeatMasker is one of such programs.
References:
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . Basic local alignment search tool. J Mol Biol 1990 Oct 5;215(3):403-10
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997 Sep 1;25(17):3389-402
Burke, J., D. Davison, et al. (1999). “d2_cluster: A Validated Method for Clustering EST and Full-Length cDNA Sequences.” Genome Res 9(11): 1135-1142.
Gotoh, O. (1982). “An improved algorithm for matching biological sequences.” J. Mol. Biol. 162: 705-708.
Green, P. (1999). SWAT/Crossmatch/PHRAP package, The University of Washington.
Gusfield, Dan. (1997). Algorithms On Strings, Trees, And Sequences: Computer Science And Computational Biology. Cambridge University Press, Cambridge, UK.
Huang, X. and A. Madan (1999). “CAP3: A DNA sequence assembly program.” Genome Res 9(9): 868-77.
Loredo J.T. (1990). From Laplace to Supernova SN 1987A: Bayesian inference in astrophysics. IN: P.F.Fougere (Ed.). Maximum Entropy and Bayesian Methods. Kluwer Academic Publishers; Dordrecht, The Netherlands. p 81-142.
Miller, R., W. Hide, et al. (1999). ISBM Tutorial No.6. EST Clustering.
Miller, R. T., A. G. Christoffels, et al. (1999). “A comprehensive approach to clustering of expressed human gene sequence: the sequence tag alignment and consensus knowledge base.” Genome Res 9(11): 1143-55.
Needleman, S. B. and C. Wunsch (1970). “A general method applicable to the search for similarities in the amino acid sequence of two proteins.” J. Mol. Biol 48: 443-453.
Ostell, J. M. and J. A. Kans (1998). The NCBI Data Model. Bioinformatics: A practical Guide to the Analysis of Genes and Proteins. A. D. Baxevanis and B. F. F. Ouellette, John Wiley & Sons, Inc: 121-144.
Pearson WR, Lipman DJ. (1988). Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A. Apr;85(8):2444-2448
Quackenbush, J., F. Liang, et al. (2000). “The TIGR Gene Indices: reconstruction and representation of expressed gene sequences.” Nucleic Acids Res 28(1): 141-145.
Schuler, G. D., M. S. Boguski, et al. (1996). “A gene map of the human genome.” Science 274(5287): 540-6.
Schuler, G. D. (1998). Sequence Alignment and Database searching. In: Bioinformatics: A practical Guide to the Analysis of Genes and Proteins. A. D. Baxevanis and B. F. F. Ouellette, John Wiley & Sons, Inc: 145-171.
Smith, T. F. and M. S. Waterman (1981). “Identification of common molecular subsequences.” J Mol Biol 147(1): 195-7.
Sutton, G. G., O. White, et al. (1995). “TIGR Assembler: a new tool for assembling large shotgun sequencing projects.” Genome Science & Technology 1(1): 9-19.
Williams. G. Nucleic Acid and Protein Sequence Databases. In: Genetic Databases. Edited by Martin J. Bishop. 1999. Academic Press, London.